Written by: Katherine Eaton
Little Hitchhikers
Humans are more than just singular beings, we are “Superorganisms”: vessels for thousands of small life forms that make up our microbiome. We have complex relationships with these resident microbes, ranging from beneficial to parasitic, which are influenced by numerous biosocial factors (diet, environment, genetics, antibiotic use, etc.). Exploring the effect these microorganisms have upon us is a hot topic of research, especially here at McMaster, as it is apparent that the microbiome plays a major role in health and disease, both physically and mentally.
But my research doesn’t seek to reinvent health care approaches. Instead, I exploit the human-microbe relationship to tell stories about how humans have migrated and exchanged diseases throughout history. And sometimes, the little microbes that have hitchhiked with us across the globe are even better storytellers than humans themselves.
Human History by Proxy
As early humans dispersed throughout the globe, human populations became geographically separate and diversified. Simultaneously, our microbiomes were co-evolving along with their human hosts resulting in the distinct geographic distributions of disease we see today. The stomach bug, Helicobacter pylori, is present in most human populations and its parallel evolution with humans has been used to reconstruct ancient patterns of human migration, dating all the way back to the dispersal out-of-Africa (Figure 1). Microbe evolution can also tell us about recent disease dispersal, such as genetically tracking the 2010 Haitian cholera epidemic back to the arrival of UN Nepalese peacekeepers. Bacterial epidemiology can also reveal altered human-environment interactions as the increased prevalence of zoonotic diseases like malaria and plague can be linked to ecological instability (ex. extensive deforestation). Microbial evolution serves as a very unique and powerful line of evidence, especially when contextual information (such as historical records) are sparse or inaccurate.
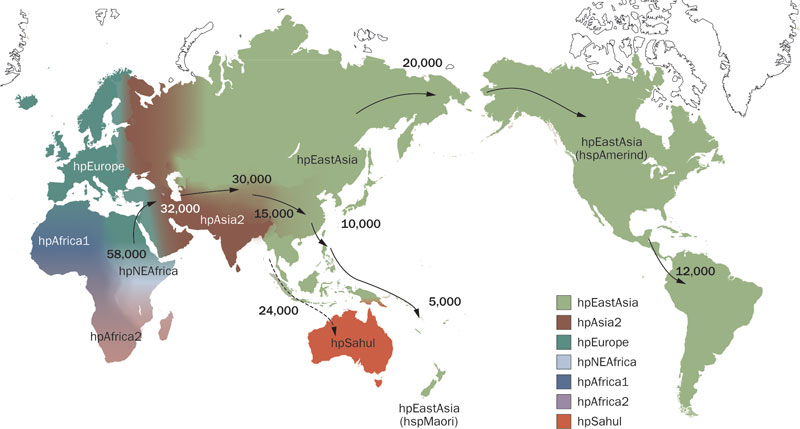
Figure 1. Helicobacter pylori global dispersal (Yamaoka 2010).
So why not just sequence human DNA rather than using a microbial proxy? Humans evolve relatively slowly (a much longer generation time) and don’t accrue as many DNA mutations within short time frames. In comparison, bacteria replicate extraordinarily quickly, with generation times ranging from 30 minutes to several hours. That means new mutations are occurring constantly, generating new data points for us to assess biological relationships, and potentially acquire finer resolution. In my case, I’m interested in both the movement of humans and how they exchanged infectious diseases in the past, therefore I turn to both bacterial DNA and historical records to reconstruct these processes.
Plague and Phylogeography
For my doctoral research, I’m examining one of humanities deadliest and ancient diseases: plague. This infectious disease is infamous for playing a major role in historical pandemics such as the Roman Plague of Justinian and the Medieval Black Death, with 30-50% of human populations perishing during these outbreaks. While mortality estimates like these have yet to be observed in the modern era, plague is still entrenched in many geographical regions in the world with the most topical being the ongoing Madagascar Plague outbreak of 2017.
Plague’s tendency to appear within a population, seemingly out of nowhere, and vanish in an equally mysterious fashion has long intrigued and frustrated researchers. Despite more than a century of comprehensive research, the origins and global routes of plague spread remain obscure. This is in part because plague is an ecologically complex disease, most commonly spread via infected rodents and their fleas, but can also spread directly between humans. In addition, the limited explanatory power of current models has also been attributed to the sparseness and ambiguity of historical mortality records, leading to high levels of uncertainty. In response, alternative lines of evidence have been cleverly sought out. Furthermore, current trends promoting the creation of open access, digital databases have greatly facilitated cross-disciplinary work and opened up previously inaccessible geographic regions and time periods for exploration.
My Project
My research continues this trend of novel lines of evidence by analyzing the ancient DNA (aDNA) of the plague bacterium in order to reconstruct disease dispersal events in human history. By extracting plague DNA from archaeological remains found in epidemic cemeteries, it is possible to identify key genetic mutations that link related bacterial strains and infer distinct waves of infection. These projects encompass a “phylogeographic” approach, which integrates phylogenetic (evolutionary) relationships with geographical relationships in order to reconstruct the spread of this infectious pathogen. The extraction, sequencing, and evolutionary analysis of plague aDNA is currently being undertaken at the McMaster Ancient DNA Centre.
The project being conducted in collaboration with the Sherman Centre for Digital Scholarship, aims to put the “geography” in the “phylogeography” of plague. Informative geospatial analysis of past pandemics is highly dependent on having strong foundational information on modern pandemics: a foundation that currently does not exist within plague literature. This is not for lack of data, as over 600 strains of plague have been sequenced and are publicly available through digital databases. This focused project therefore aims to curate, contextualize, and analyze the digital metadata associated with these plague strains. Armed with this comparative data, I will then be able to start exploring hypotheses such as:
- Did trade routes and migration events influence the distribution of plague?
- Are there ecological zones that correlate with increased prevalence of plague?
- Does genetic evidence complement or contradict archival-based models of plague spread?
My objective is to expand our epidemiological knowledge of plague, in a way that improves our understanding of the interplay of factors contributing to modern re-emergences, as well as the historical events that triggered past pandemics. The combination of molecular genetics and geospatial analysis, driven by humanities-focused questions offers a unique lens through which to reconstruct the fluctuating patterns of human connectivity and ecological interaction that have shaped our relationship with infectious disease.
Project Organization: The Beginning of the End?
When I’m trying to organize a project, my favorite place to start is… at the end. What kind of finished product do I want to end up with? And how might the answers to these questions be visualized and explored? There’s no shortage of geospatial tools to explore disease epidemiology, so I created 5 criteria to assist in program selection:
- Statistical Framework – Hypothesis testing is a must, visualization alone is insufficient.
- Disparate Data – Incorporate heterogeneous metadata and account for prior information about evolutionary relationships. (Bayesian GIS anyone?)
- Standardized Output – Produce an output file that will be recognized by other geospatial and visualization software. Proprietary file formats are to be avoided.
- Aesthetics – Try to avoid the Google Maps API (personal preference).
- Learning Barriers – Free, open source, reduced learning curve or plentiful training resources.
As expected, no program satisfies all 5 criteria, and thus I will likely use a combination to highlight each tool’s strengths and complement weaknesses. There are a variety of R packages that seem promising, the best candidate being BPEC (Bayesian Phylogeographic and Ecological Clustering) (Figure 2). This tool has powerful analytical potential and can identify meaningful geographic clusters in your data. To go deeper into routes of spread, SpreaD3 is well-designed for epidemic source tracking and takes as input files from programs which I am already using (Figure 3). With a faint idea of what I need to prepare for downstream, I was better prepared to select appropriate datasets.
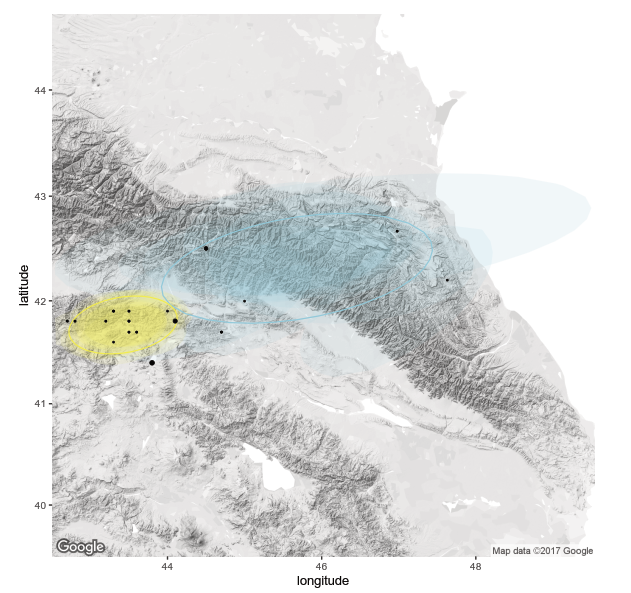
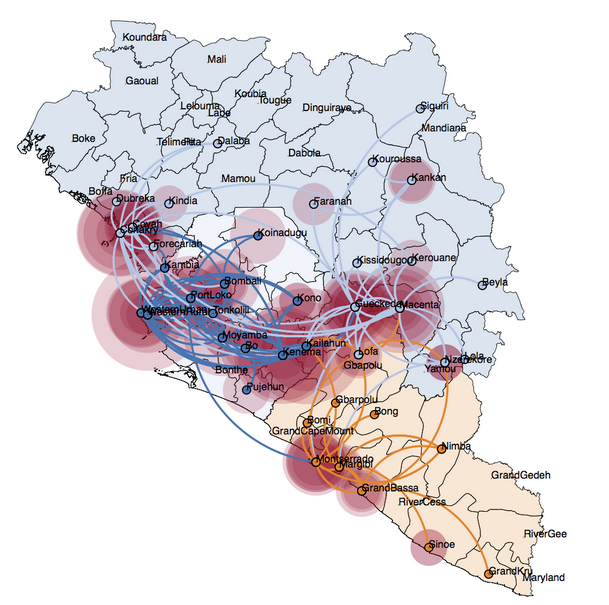
Project Organization: Let’s Get Started
For the previously identified programs, there are three mandatory pieces of information I need for each outbreak record:
- DNA sequence data to reconstruct evolutionary relationships.
- Geographic location (ideally latitude and longitude).
- Collection year (time point calibration).
Accessory variables that would be very interesting to test include host (rodent, human, camel, etc.) but are rarely made available by submitters.
Based on my review of plague literature, I was expecting to find about 150 plague genome records as this number seemed to be representative of current publications. I began my search scouring online genome repositories (NCBI, ENA, DDBJ) to identify datasets. To my great surprise, I found over 600 plague genome sequencing projects which either had been 1) published on but quality could be improved, 2) published on but only in a limited descriptive sense, or 3) has no publications associated with it. Despite being overwhelmed with an unexpected amount of data, I’m still very excited by the potential to contribute something new and meaningful with data that is mostly untouched.
The problem is that this data sits behind a very scary wall: big data science. The actual genetic sequence data is enormous and complex (we’ll save that for another post) and the metadata is messy with many missing fields. The messiness can in part be cleaned up with tools like OpenRefine, but the missing data either means I’m going manually hunting through Supplementary Files, or a lot of data is getting kicked out.
I then investigated how to query, download, and parse over 600 files of metadata into a meaningful table I could import into downstream applications. I tested out a number of APIs (Bioconductor, Biopython, SRAdb, MetaSRA) but wound up dissatisfied in part with most of them. My current strategy is rather ad-hoc, as I intend to combine multiple programmatic APIs, web-browser GUIs, and my own python scripts to build up a geospatial relational database. The result is functional, but unwieldy, and is rather limited in application to my own project (Figure 4). I’m also currently missing about 300 database records, largely because of consistency issues inherent to repositories that are governed by user-submission.
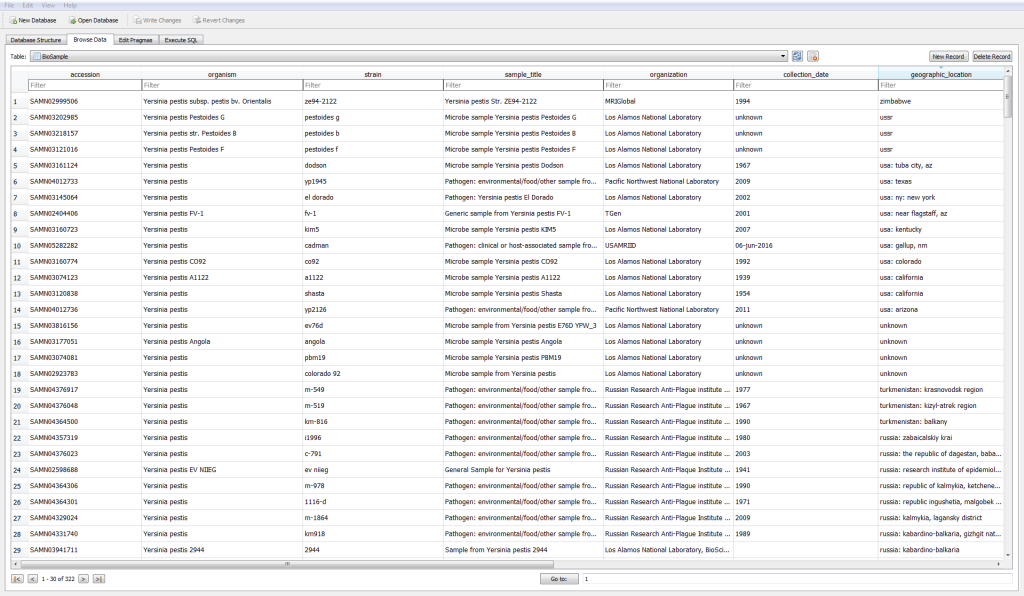
Figure 4. One table (among many) present in my SQLite relationship database.
Next Steps
My next goal is to get the database pipeline to a point where I’m satisfied it has scraped as much information as it can via automation. From there I will move to manually going through associated publications to fill in geographic location and date where I can. At that point, it will be time to start experimenting with geospatial tools to figure out how I’m going to visualize hundreds of years of global disease dispersal in an informative manner. I’m looking forward to geospatial workshops in the new year, and I’ll be showcasing some preliminary maps next blog post!
Figure Reference
Bielejec, F., Baele, G., Vrancken, B., Suchard, M. A., Rambaut, A., Lemey, P. (2016) SpreaD3: interactive visualisation of spatiotemporal history and trait evolutionary processes. Molecular Biology and Evolution. 33 (8): 2167-2169.
Manolopoulou, I., Hille, A., Emerson, B. (2017). BPEC: An R package for bayesian phylogeographic and ecological clustering. Journal of Statistical Software. arXiv:1604.01617v2
Yamaoka, Y. (2010). Mechanisms of disease: Helicobacter pylori virulence factors. Nature Reviews Gastroenterology & Hepatology. 7: 629–641

Leave a Reply