Written by: Katherine Eaton
In my last residency update, I discussed two major goals:
- Reconstructing the spread of a historically important disease (the Plague!), and
- Creating a digital exhibit combining interactive visuals with narrative text.
I’m pleased to say that I have a working demo of the Plague Exhibit which is hosted through the NextStrain community page. However, I worry that the deployed exhibit will be quite fragile for now as I continue testing and development. I’d like to use this blog post to contextualize the process and findings a little bit more.
The data used in this demo are 151 samples of the plague bacterium Yersinia pestis which were submitted for genome sequencing and made publicly accessible through the National Centre for Biotechnology Information. There are many more samples publicly available, I simply selected those with the best available metadata to construct my demo exhibit.
The computational methods used for this demo are part of my Plague GitHub Repository, which are still highly experimental in nature. I’ve been working on this analytical pipeline since 2017! However, the methods changed dramatically in 2019-2020 in part because of the research response to COVID-19 which emphasized the collaborative sharing of knowledge in real-time.
The purpose of creating this demo exhibit is to perform a spread reconstruction… and then immediately critique it and tear it apart. I’ve found that interactive visuals spark inventive questions which often lead to productive conversations.
Geographic Spread
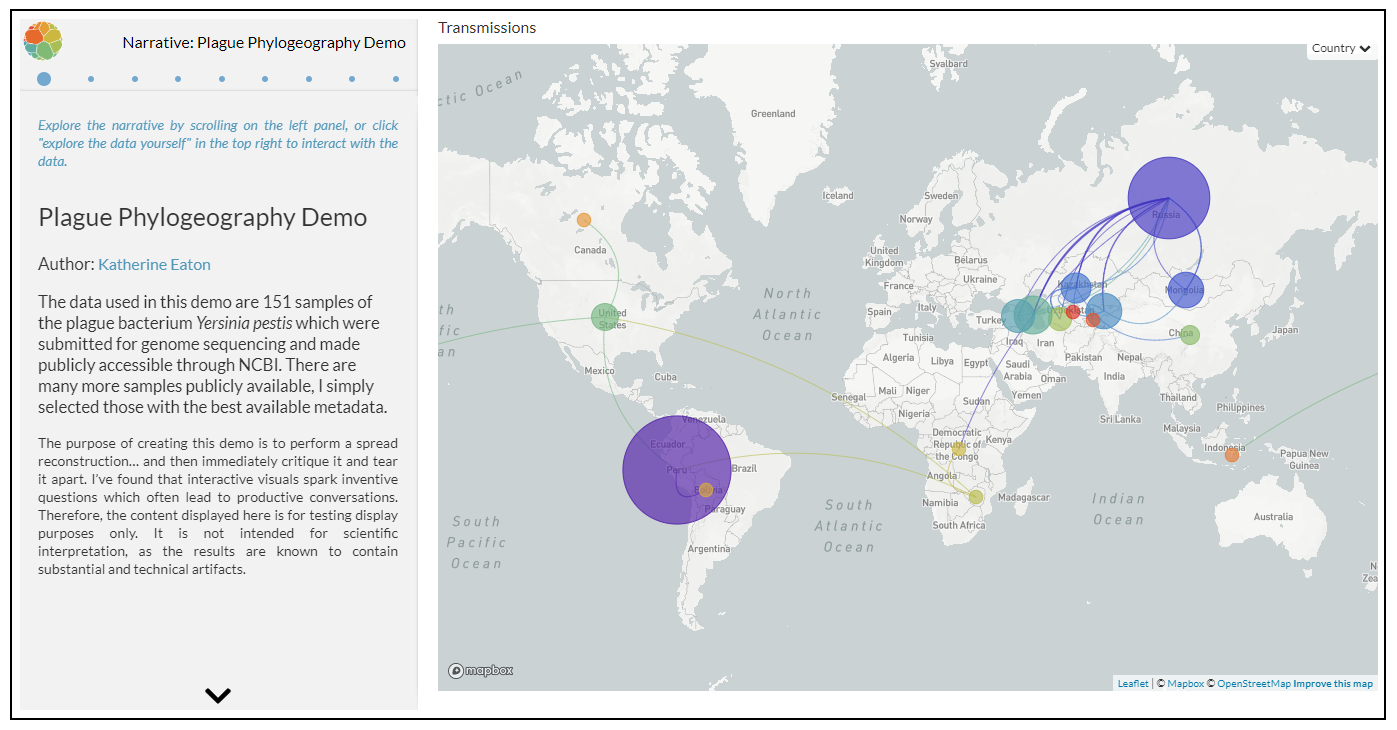
The following visual is a spread reconstruction of the plague bacterium Y. pestis across the globe. It’s constructed by examining an evolutionary tree, where the geographic location of plague samples at the tips/leaves are known, and locations in the past are statistically inferred.
This animation is congruent with the hypothesis that plague’s origins are in Asia, possible from Russia, China, Mongolia, etc. However, we see a cardinal sin of geocoding in action, as I’ve simply used the sample’s “Country” metadata field to retrieve latitude and longitude which is now being placed in the centroid of each country. Russia is an incredibly large landmass, and the nuances of sampling location WILL hugely bias the spread reconstruction.
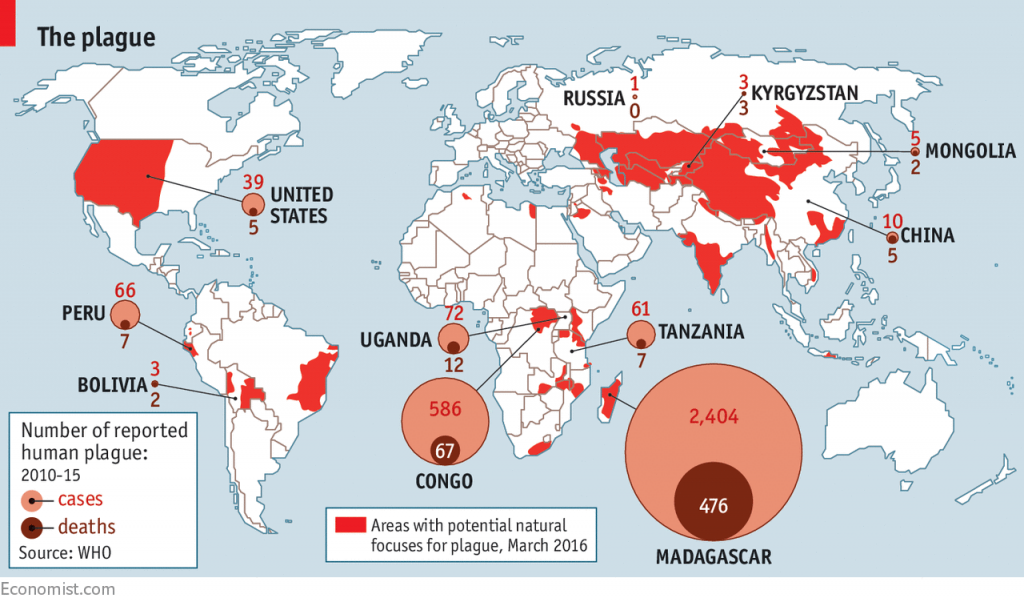
When the animation finishes, compare the locations of the largest bubbles in the figure above to the location of the largest bubbles in the figure below. Why are they so different? Admittedly, my own dataset is a small and biased sample. Increasing my sample size will yield additional samples from Madagascar but frustratingly little more from the DRC, Tanzania, or Uganda, the most heavily affected places today. It is extremely concerning to know areas of high public health importance are so severely underrepresented in narratives constructed from genetic material.
Plague’s Not Picky
Another reason those maps differ is because the plague is not picky, the disease can infect virtually all mammals. In the following evolutionary tree, the bubbles (plague samples) are coloured by the host they were collected from. Frustratingly, no clear patterns of host clusters emerge, the colours appear randomly distributed. I suspect there are far more ecologically meaningful ways to colour/label the hosts including: domestic vs. non-domestic species, reservoir vs. non-reservoir species etc.
The Issue of Time…
While the geographic origins of the spread reconstruction made sense, there are many reasons to be suspicious. Accurately modelling these dynamics requires several criteria to be met. The most important one being the presence of temporal signal or more plainly, a correlation between how much time has passed and how many mutations have occurred.
The following plot explores exactly that: by regression of time (date on the X-axis) against mutations (genetic divergence Y-axis). The plague samples (coloured dots) are widely dispersed above and below the regression line (black line), a highly undesirable result. The finding that some plague strains evolve slower or faster than others is widely known, and it continues to be a thorny problem!
Where’s the “Human”ities?
To wrap things up, I’ll talk briefly about the “human” side of the project. The following map shows how human outbreaks of plague occur across various continents:
The following tree highlights plague samples isolated from human hosts. Rather than clustering all within one part of the tree, this again shows plague’s fluidity to move between hosts as many strains are capable of infecting humans.
What’s Next?
With a successful demo deployed, I’m looking forward to doing some serious scientific inquiry! My next steps will be to expand my sample size to include more datasets that, while previously published, have never been synthesized and visualized in this way before. Following that, I’ll be asking these questions:
- How has the composition of plague data changed in the last 10 years?
- Does a compositional bias (ex. geography) change our historical narratives of disease?
- What are the dangers in synthesizing digital datasets created from disparate agendas and objectives? What are the possible contributions that can be made?

Leave a Reply